Pęcherzowe oddzielanie się naskórka Spis treści Epidemiologia | Etiopatogeneza i klasyfikacja | EB...
Choroby skóryChoroby genetyczneRzadkie choroby
łac.pęcherzowychskórygenodermatozkeratynęBłony śluzowemikroskopie elektronowymcytoplazmatyczneBliznowacenieprosakówbłon śluzowychpłytkowatągorączkakeratynyautosomalnie recesywnielamininynadżerkidróg oddechowychbliznowaceniesyndaktyliaprosakistrupsepsaautosomalne recesywneatrezjaodźwiernikaatrezja przełykudwunastnicyhipoplazjaszkliwagrudkowychdystroficznekolagenpseudosyndaktyliazwężenie przełykustulejkaraka kolczystokomórkowego skóryZapoznaj się z zastrzeżeniami dotyczącymi pojęć medycznych i pokrewnych w Wikipedii
| Pęcherzowe oddzielanie naskórka | |||||||||||||
epidermolysis bullosa | |||||||||||||
| |||||||||||||
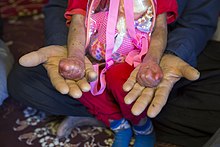
Pęcherzowe oddzielanie się naskórka (łac. epidermolysis bullosa) – grupa pęcherzowych chorób skóry uwarunkowanych genetycznie (genodermatoz). Wspólną cechą tych chorób jest występowanie pęcherzy wskutek urazów mechanicznych.
Spis treści
1 Epidemiologia
2 Etiopatogeneza i klasyfikacja
3 EB epidermolytica (EB simplex, EBS)
4 EB junctionalis (EBJ)
5 Postać dystroficzna (EB dystrophica)
6 Charakterystyka poszczególnych odmian EB
6.1 Grupa epidermolysis bullosa simplex
6.1.1 EB Webera-Cockayne’a
6.1.2 EB Dowlinga-Meary (EB herpetiformis)
6.1.3 EB wariant Koebnera
6.2 Grupa epidermolysis bullosa junctionalis
6.2.1 EB Herlitza
6.2.2 EB z atrezją odźwiernika
6.3 Grupa epidermolysis bullosa dystrophica
6.3.1 EB dystroficzna, postać Pasiniego
6.3.2 EB dystroficzna, postać Cockayne’a-Toureine’a
6.3.3 EB Hallopeau-Siemensa
7 Zobacz też
8 Przypisy
9 Bibliografia
10 Linki zewnętrzne
Epidemiologia |
Choroby grupy epidermolysis bullosa są niezwykle rzadkie. Szacuje się, że występują z częstością 2/100 000 osób.
Etiopatogeneza i klasyfikacja |
Wyróżnia się trzy główne grupy, w zależności od miejsca tworzenia się pęcherza:
- w obrębie naskórka (EB simplex)
- w obrębie błony podstawnej (EB junctionalis)
- poniżej błony podstawnej, w skórze właściwej (EB epidermolytica).
Postęp w biologii molekularnej umożliwił niedawno dermatologom na ujednolicenie klasyfikacji EB, co doprowadziło do konsensusu w kwestii nazewnictwa i podziału.
| Grupa | Typy | Uszkodzony gen | Nieprawidłowe białko | OMIM |
|---|---|---|---|---|
| EB simplex (EBS) | EBS Weber-Cockayne EBS Koebner EBS Dowling-Meara EBS z dystrofią mięśniową | KRT 5 i KRT 14 KRT 5 i KRT 14 KRT 5 i KRT 14 PLEC 1 | Keratyna 5 i 14 Keratyna 5 i 14 Keratyna 5 i 14 Plektyna | OMIM 131800 OMIM 131900 OMIM 131760 OMIM 226670 |
| EB junctionalis (EBJ) | EBJ Herlitz EBJ non-Herlitz EBJ z atrezją odźwiernika | LAMB3, LAMC2, LAMA3 COL17A1, LAMB3, LAMC2, LAMA3 ITGA6, ITGB4 | Laminina 5 Kolagen XVII, laminina 5 α6 β4 Integryna | OMIM 226700
|
| EB dystrophica (EBD) | EBD Hallopeau-Siemens EBD non-Hallopeau-Siemens EBD dziedziczona autosomalnie dominująco | COL7A1 COL7A1 COL7A1 | Kolagen VII Kolagen VII Kolagen VII | OMIM 226600 OMIM OMIM 131800 |
EB epidermolytica (EB simplex, EBS) |
W tej grupie wyróżnia się:
- odmianę poronną Webera-Cockayne’a postaci zwykłej EB (EBS-WC) OMIM 131800
- odmianę Koebnera postaci zwykłej EB (EBS-K, EBS2) OMIM 131900
- odmianę Dowlinga-Meary postaci zwykłej EB (EBS-DM) OMIM 131760
- EBS z cętkowaną pigmentacją (EB with mottled pigmentation; EBS-MP) OMIM 131960
- odmianę z dystrofią mięśniową postaci zwykłej EB (EB simplex and limb-girdle muscular dystrophy MD-EBS; MDEBS, EB-MD) OMIM 226670
- odmiana Ogna (EBSO, EBS1) OMIM 131950
- EB simplex dziedziczona autosomalnie recesywnie OMIM 601001
EB junctionalis (EBJ) |
- odmiana Herlitza (letalna) EBJ (odmiana Herlitza-Pearsona, epidermolysis bullosa letalis, EBJ) OMIM 226700
- odmiana ‘non-Herlitz’ EBJ (EB, generalized atrophic benign, GABEB) OMIM 226650
- odmiana EBJ z atrezją odźwiernika, zespół Carmiego (EB with pyloric atresia, EB-PA, PA-JEB) OMIM 226730
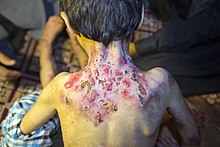
Postać dystroficzna (EB dystrophica) |
W obrębie tej podgrupy wyróżnia się następujące jednostki chorobowe:
- postać dystroficzna EB odmiana Hallopeau-Siemensa (EB dystrophica, Hallopeau-Siemens type; severe RDEB); EBR1 OMIM 226600
- postać dystroficzna EB odmiana ‘non-Hallopeau-Siemens’ (RDEB mitis)
- postać dystroficzna EB odmiana dziedziczona autosomalnie dominująco
- postać dystroficzna wariant Pasiniego (EB dystrophica, Pasini type, allopoliploid dominant dystrophic EB; EBDD OMIM 131750)
- postać dystroficzna wariant Cockayne’a-Toureine’a (EB dystrophica, Cockayne-Touraine type) OMIM 131800
- postać Barta (EB with congenital localized absence of skin and deformity of nails, EB dystrophica, Bart type) OMIM 132000
Charakterystyka poszczególnych odmian EB |
Grupa epidermolysis bullosa simplex |
EB Webera-Cockayne’a |
Choroba spowodowana jest mutacją w genie KRT14 kodującego keratynę 5 i 14 oraz defektem enzymów proteolitycznych. Dziedziczona jest autosomalnie dominująco[1]. Zmiany występują najczęściej na stopach, rzadziej na dłoniach; mogą jednak pojawić się w każdej lokalizacji. Przebieg jest stosunkowo łagodny. Pęcherze rzadko obecne są już przy urodzeniu; zwykle pojawiają się u dzieci w wieku około 18 miesięcy, gdy wykazują dużą aktywność ruchową. Zmiany mogą się ujawnić w późniejszym wieku, w późnym dzieciństwie albo u młodych dorosłych. Klasyczne są przypadki młodych rekrutów u których choroba ujawniła się po pierwszym forsownym marszu. Czynniki nasilające tworzenie pęcherzy to duża wilgotność, wzmożone pocenie się. Błony śluzowe i płytki paznokciowe nie są zajęte.
EB Dowlinga-Meary (EB herpetiformis) |
Od innych chorób z grupy epidermolysis bullosa simplex różni się ciężkim przebiegiem i charakterystycznym obrazem w mikroskopie elektronowym (cytoplazmatyczne „kępki” tonofilamentów). Tak jak typ Koebnera i typ Webera-Cockayne’a, typ Dowlinga-Meary EBS wiąże się z mutacjami w genach KRT5 (region 12q13.13) i KRT14 (region 17q21.2)[2]. Objawy[3]: uogólnione tworzenie pęcherzy o opryszczkowatym (zgrupowanym) układzie, stąd inna nazwa schorzenia, epidermolysis bullosa simplex herpetiformis. Surowicze i krwotoczne pęcherze tworzą się w każdej lokalizacji, przede wszystkim jednak na stopach i podeszwach, dookoła ust, na karku i szyi. Bliznowacenie nie jest regułą, ale proces zapalny, zwłaszcza wikłający pęcherze krwotoczne, może skutkować tworzeniem blizn i prosaków. Notowano zajęcie błon śluzowych i płytkowatą hiperkeratozę dłoni i podeszew. Podpaznokciowe tworzenie pęcherzy może powodować utratę paznokci. Czynnikami zaostrzającymi przebieg choroby są: gorączka, pora roku (miesiące letnie), a także wiek (zmiany nasilają się z wiekiem).
EB wariant Koebnera |
Choroba spowodowana jest mutacjami w genach keratyny 5 i 14 (KRT4, KRT15). Ponieważ mutacje w tych samych genach odpowiadają za wystąpienie cech fenotypowych EBS typu Webera-Cockayne’a i typu Dowlinga-Meary, często cechy tych trzech chorób nakładają się na siebie. Zdarza się, że w obrębie jednej rodziny diagnozowano przypadki dwóch różnych typów EBS. Zmiany są hiperkeratotyczne, pęcherzowe, czasem pęcherze są krwotoczne; w obrazie klinicznym występują nadżerki. Manifestują się już od urodzenia albo we wczesnym niemowlęctwie. Niekiedy tworzą się prosaki, mogą wystąpić zaburzenia pigmentacji (hipo- lub hiperpigmentacja) tak jak w odmianie Dowlinga-Meary, czasem zajęte są paznokcie i błony śluzowe.
Grupa epidermolysis bullosa junctionalis |
EB Herlitza |
Postać śmiertelna, rzadka. Dziedziczona jest autosomalnie recesywnie. Spowodowana jest mutacją w jednym z genów kodujących trzy polipeptydy lamininy 5: α-3 (gen LAMA3), β-3 (gen LAMB3), albo γ-2 (gen LAMC2)[4]. U noworodków od urodzenia występują liczne pęcherze, które łatwo pękają, pozostawiając rozległe nadżerki w obrębie całej skóry i błon śluzowych, również dróg oddechowych. Już wewnątrzmacicznie następuje tworzenie pęcherzy i nadżerek, tak że dziecko rodzi się z rozległymi ubytkami naskórka. Ręce i stopy są zwykle relatywnie najmniej dotknięte przez proces chorobowy. Nigdy nie występuje bliznowacenie, syndaktylia, nie tworzą się prosaki. W wieku około 6 miesięcy wokół ust bądź nosa tworzy się patognomoniczny, niegojący się strup. Zgon następuje w okresie niemowlęcym mimo intensywnej opieki medycznej, stosowania antybiotyków, kortykosteroidów, wyrównania poziomu elektrolitów. Przyczyną śmierci jest utrata białka surowicy i sepsa. W ostatnich latach podejmowano próby przeszczepiania skóry[5].
EB z atrezją odźwiernika |
Bardzo rzadka choroba spowodowana mutacją w genach ITGB4 i ITGA6[6]. W skórze chorych można stwierdzić obniżoną ekspresję kodowanych przez te geny α6 i (lub) β4 integryny[7]. Dziedziczenie jest autosomalne recesywne. Objawy są obecne przy urodzeniu albo pojawiają się we wczesnym niemowlęctwie. Zmianom skórnym z reguły towarzyszy wrodzona atrezja odźwiernika (rzadziej atrezja przełyku lub dwunastnicy). Często występuje dystrofia paznokci i anomalie w zakresie uzębienia, takie jak hipoplazja szkliwa. Tworzenie pęcherzy jest uogólnione, rokowanie bardzo złe, mimo chirurgicznej interwencji; mutacja często jest letalna. W wariantach letalnych (zwykle spowodowanych mutacją typu nonsens) zgon następuje wkrótce po urodzeniu; w pozostałych przypadkach obserwuje się tendencję do zaostrzania zmian skórnych z wiekiem.
Grupa epidermolysis bullosa dystrophica |
EB dystroficzna, postać Pasiniego |
Dziedziczona jest w sposób autosomalny dominujący. Pęcherze ustępują z pozostawieniem drobnych, białawych wykwitów grudkowych, umiejscowionych głównie na tułowiu w okolicy krzyżowej. Zajęte są śluzówka jamy ustnej i zęby. Paznokcie mogą być dystroficzne. W starszym wieku choroba może złagodzić swój przebieg.
EB dystroficzna, postać Cockayne’a-Toureine’a |
Dziedziczona jest w sposób autosomalny dominujący. Wykwity głównie na kończynach, śluzówka i zęby zajęte w minimalnym stopniu[8].
EB Hallopeau-Siemensa |
Choroba wywołana jest mutacją w genie kodującym kolagen typu VII (COL7A1). Charakterystyczne jest uogólnione tworzenie pęcherzy w momencie urodzenia lub we wczesnym niemowlęctwie, rozległe bliznowacenie, zmiany w obrębie skóry kończyn. Miejsca mające predylekcję do pęcherzy to dłonie, stopy, łokcie i kolana. Może wystąpić pseudosyndaktylia dłoni i stóp, niekiedy są zajęte paznokcie i zęby, śluzówka przełyku (czego skutkiem jest zwężenie przełyku), stenoza cewki moczowej, odbytu, stulejka, bliznowacenie rogówki lub spojówki. W EB Hallopeau-Siemensa wyraźnie zwiększone jest ryzyko wystąpienia raka kolczystokomórkowego skóry; nowotwór rozwija się na podłożu blizn[9].
Zobacz też |
- Jonny Kennedy
Przypisy |
↑ KanaK. Yasukawa KanaK. i inni, Dominant and Recessive Compound Heterozygous Mutations in Epidermolysis Bullosa Simplex Demonstrate the Role of the Stutter Region in Keratin Intermediate Filament Assembly, „Journal of Biological Chemistry”, 26, 2002, s. 23670–23674, DOI: 10.1074/jbc.M200974200, ISSN 0021-9258, PMID: 11973334 [dostęp 2017-01-03] (ang.).
↑ Fine, J.-D., Eady, R. A. J., Bauer, E. A., Bauer, J. W., Bruckner-Tuderman, L., Heagerty, A., Hintner, H., Hovnanian, A., Jonkman, M. F., Leigh, I., McGrath, J. A., Mellerio, J. E., Murrell, D. F., Shimizu, H., Uitto, J., Vahlquist, A., Woodley, D., Zambruno, G. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J. Am. Acad. Derm. 58: 931-950, 2008. [PubMed: 18374450, related citations].
↑ Hacham-Zadeh, S., Rappersberger, K., Livshin, R., Konrad, K. Epidermolysis bullosa herpetiformis Dowling-Meara in a large family. J. Am. Acad. Derm. 18: 702-706, 1988. [PubMed: 3372762, related citations].
↑ F.J.D.F.J.D. Smith F.J.D.F.J.D. i inni, Plectin deficiency results in muscular dystrophy with epidermolysis bullosa, „Nature Genetics”, 4, 1996, s. 450–457, DOI: 10.1038/ng0896-450 [dostęp 2017-01-03] (ang.).
↑ Skoczylas M., Kordek A., Łoniewska B., Maleszka R., Rudnicki J., Problemy kliniczne u chorych na odmianę Herlitza pęcherzowego oddzielania się naskórka. Postępy Neonatologii 2011;2(17):54-56.
↑ FrédériqueF. Vidal FrédériqueF. i inni, Integrin β4 mutations associated with junctional epidermolysis bullosa with pyloric atresia, „Nature Genetics”, 2, 1995, s. 229–234, DOI: 10.1038/ng0695-229 [dostęp 2017-01-03] (ang.).
↑ S.S. Chavanas S.S. i inni, Splicing Modulation of Integrin (β4 Pre-mRNA) Carrying a Branch Point Mutation Underlies Epidermolysis Bullosa with Pyloric Atresia Undergoing Spontaneous Amelioration With Ageing, „Human Molecular Genetics”, 11, 1999, s. 2097–2105, DOI: 10.1093/hmg/8.11.2097, ISSN 0964-6906, PMID: 10484780 [dostęp 2017-01-03] (ang.).
↑ C.A.C.A. Oakley C.A.C.A. i inni, The Cockayne-Touraine type of dominant dystrophic epidermolysis bullosa--ultrastructural similarities to the Pasini variant, „Acta Dermato-Venereologica”, 3, 1984, s. 253–256, ISSN 0001-5555, PMID: 6204490 [dostęp 2017-01-03] .1 stycznia
↑ J.J. Diedrichson J.J., D.D. Talanow D.D., A.A. Safi A.A., [Epidermolysis bullosa dystrophica (Hallopeau-Siemens syndrome) of the hand -- surgical strategy and results], „Handchirurgie, Mikrochirurgie, Plastische Chirurgie: Organ Der Deutschsprachigen Arbeitsgemeinschaft Fur Handchirurgie: Organ Der Deutschsprachigen Arbeitsgemeinschaft Fur Mikrochirurgie Der Peripheren Nerven Und Gefasse: Organ Der V...”, 5, 2005, s. 316–322, DOI: 10.1055/s-2005-872849, ISSN 0722-1819, PMID: 16287016 [dostęp 2017-01-03] .
Bibliografia |
- Stefania Jabłońska, Sławomir Majewski Choroby skóry i choroby przenoszone drogą płciową PZWL 2005, ISBN 83-200-3367-5.
- GernotG. Rassner GernotG., Dermatologia. Podręcznik i atlas, WojciechW. Silny (tłum.), Wrocław: Urban & Partner, 1994, ISBN 83-85842-25-X, ISBN 3-541-08374-3, OCLC 749381926 .

„Czerwona wstążka” – symbol stowarzyszeń chorych na epidermolysis bullosa i ich rodzin.
Linki zewnętrzne |
Epidermolysis bullosa with pyloric atresia na stronie Geneva Foundation for Medical Education and Research (ang.)
Artykuł z eMedicine (ang.)
Epidermolysis Bullosa Medical Research Foundation (ang.)
Netzwerk Epidermolysis bullosa (niem.)
Epidermolysis Bullosa Simplex w GeneReviews (ang.)
![]() Zapoznaj się z zastrzeżeniami dotyczącymi pojęć medycznych i pokrewnych w Wikipedii.
Zapoznaj się z zastrzeżeniami dotyczącymi pojęć medycznych i pokrewnych w Wikipedii.
Kontrola autorytatywna (choroba):
BNCF: 42095

