Siatkówczak Spis treści Historia | Epidemiologia | Patogeneza | Objawy i przebieg | Patomorfologia...
Artykuły na medalChoroby narządów zmysłówNowotwory dziedziczneRzadkie chorobyChoroby dziedziczone autosomalnie dominująco
łac.nowotwór złośliwyczerniakunaczyniówkimutacjebiałko retinoblastomaleukokoriazezzapadalnośćPeruLejdyPieter PauwJames WardropparyscyRudolf Virchowrozetek Flexnera-Wintersteinera1926Alfred Knudsonhipotezę dwóch uderzeńzłośliwymsiatkówkizapadalnościPolscekwasu foliowegowirusem brodawczaka ludzkiegozapłodnienie in vitroryzyko względneantyonkogenuchromosomie 13autosomalnie dominującopenetracjąmutacjiAlfreda Knudsonahipoteza dwóch uderzeńallelleukokoriazezlampą błyskowąefekt czerwonych oczurubeoza tęczówkiropostekkrwistekwolooczezapalenie tkanki łącznejwytrzeszczszyszynkiokolicy nadsiodłowejguzów drobnookrągłoniebieskokomórkowychjądrzecytoplazmyretinoblastymitotycznegomartwicyneuroblastomaznieczuleniu ogólnymrąbek zębatywziernikowaniemodwarstwienie siatkówkiciała szklistegochoroba Coatsanerwu wzrokowegochemioredukcjaChemioterapiablaszką sitowąkarboplatynawinkrystynaetopozydcyklosporynę AśródbłonkazawałpodczerwienikarboplatynęakceleratorówteleradioterapiabrachyterapiakolimatorycGyneowaskularyzacja tęczówkijodurutenuiryduneuropatiatorebkę TenonaThioTEPAkarboplatynanoża diatermicznegohydroksyapatytupolietylenuadenowirusowegoteleradioterapiachemioterapiamięsakiczerniak złośliwyraki sutkapłuclinii komórkowychterapii genowejprobantaZapoznaj się z zastrzeżeniami dotyczącymi pojęć medycznych i pokrewnych w Wikipedii
| Siatkówczak | |||||||||||||||||||||
 Leukokoria (odblask źrenicy) prawego oka wskazująca na obecność guza | |||||||||||||||||||||
| |||||||||||||||||||||
Siatkówczak (łac. retinoblastoma) – najczęstszy wewnątrzgałkowy nowotwór złośliwy oka u dzieci. Jeśli weźmie się pod uwagę wszystkie grupy wiekowe, zajmuje on drugie miejsce po czerniaku naczyniówki w tej grupie nowotworów. Rozwój nowotworu jest inicjowany przez mutacje, które dezaktywują obie kopie genu RB1, kodującego białko retinoblastoma. Pierwszym objawem choroby jest najczęściej leukokoria – pojawienie się białego odblasku w oku lub obu oczach, albo zez. Siatkówczak jest rzadką chorobą – ocenia się, że zapadalność na ten typ nowotworu wynosi około 4 przypadków na milion na rok (w latach 1990-1995, wśród dzieci poniżej 15. rż.)[1].
Guz w 2/3 przypadków rozwija się w jednej gałce ocznej, w 1/3 w obu (ale zwykle niejednocześnie). Choroba może mieć charakter dziedziczny lub niedziedziczny.
Siatkówczak występuje prawie wyłącznie u dzieci poniżej 5. roku życia i stanowi około 3% nowotworów złośliwych występujących u dzieci poniżej 15. roku życia. Przypadki u dorosłych również zostały odnotowane, aczkolwiek są niezmiernie rzadkie.
Spis treści
1 Historia
2 Epidemiologia
3 Patogeneza
4 Objawy i przebieg
5 Patomorfologia
5.1 Histopatologiczne czynniki ryzyka
6 Rozpoznanie
7 Testy genetyczne
8 Różnicowanie
9 Leczenie
9.1 Leczenie ogólnoustrojowe i chemioredukcja
9.2 Leczenie miejscowe
9.3 Terapie eksperymentalne
9.4 Regresja guza
10 Rokowanie
11 Nowe kierunki badań
12 Poradnictwo genetyczne i profilaktyka
13 Uwagi
14 Przypisy
15 Linki zewnętrzne
Historia |

Fragment pracy Pietera Pauwa, przedrukowanej pośmiertnie przez Thomasa Bartholina[2]. Obok ryciny autorstwa Andreasa Stoga przedstawiającej Pauwa dokonującego publicznej sekcji w tamtejszym teatrze anatomicznym znajduje się tekst jego XXIII obserwacji dotyczącej guza oka u 3-letniego chłopca

Ilustracja z pracy Wardropa z 1809 roku przedstawiająca guz oka u dziecka[3]
Przypuszczalnie najstarszym dowodem występowania tej choroby jest rzeźba z Peru pochodząca sprzed 2000 lat[4]. Pierwszy opis prawdopodobnie dotyczący siatkówczaka pozostawił profesor anatomii i botanik z Lejdy Pieter Pauw[5], na co zwrócił uwagę w 1963 roku Edwin B. Dunphy[6]. Opis Pauwa odpowiada obrazowi klinicznemu nieleczonego siatkówczaka; guz zajmował oczodół, okolicę skroniową i czaszkę. Tkanka guza miała przypominać "tkankę mózgową zmieszaną z gęstą krwią i pokruszonymi kamieniami"[2]. W 1805 roku William Hey utworzył termin fungus haematodes, którym określił grzybiastą masę guza zajmującego gałkę oczną i niszczącego jego wewnętrzną strukturę. W 1809 szkocki chirurg James Wardrop zebrał obserwacje innych autorów[3]; mimo że nie dysponował mikroskopem, skrupulatne badania pozwoliły mu stwierdzić, że w większości przypadków guz wywodził się z siatkówki. Wardrop udokumentował szerzenie się guza na nerw wzrokowy i mózg, a później opisał przerzuty siatkówczaka do różnych części ciała. W 1836 roku paryscy badacze Langenbech, Robin i Nystin potwierdzili histologicznie pochodzenie guza z tkanki siatkówki.
W 1864 roku Rudolf Virchow nazwał ten typ guza glejakiem siatkówki (glioma retinae) wskazując komórki glejowe siatkówki, na te, z których wywodzi się guz[7].
W 1891 Flexner ze szpitala Johnsa Hopkinsa pierwszy zauważył rozetki, w które układają się komórki guza[8]. Kilka lat później, w 1897, Wintersteiner[9] potwierdził obserwacje Flexnera i zaproponował termin neuroepithelioma zauważając podobieństwo komórek guza do pręcików i czopków siatkówki. Od nazwisk dwóch badaczy pochodzi eponim rozetek Flexnera-Wintersteinera.
Veorhoff[10] był pierwszym który stwierdził, że nowotwór wywodzi się z macierzystych komórek siatkówki i wprowadził termin retinoblastoma, który w 1926 roku Amerykańskie Towarzystwo Oftalmologiczne uznało za obowiązujący.
Ts'o i wsp. w 1970 ostatecznie ustalili, że guz wywodzi się z komórek prekursorowych fotoreceptorów[11].
Pierwszy dowód na zmiany cytogenetyczne w komórkach guza przedstawiono w latach 60., gdy kariotypowanie pozwoliło stwierdzić delecję jednego z chromosomów grupy D[12][13]. W 1971 roku Orye opisał delecję 13q w dwustronnym siatkówczaku[14]. W tym samym roku Alfred Knudson przedstawił swoją hipotezę dwóch uderzeń ("two hits"), tłumaczącą genetyczne podstawy nowotworzenia na przykładzie siatkówczaka[15].
Historia leczenia siatkówczaka liczy około stu lat. W 1903 roku Hilgartner opisał pierwszy przypadek wyleczenia dziecka z guzem przy pomocy radioterapii[16]. Od tego momentu przeżywalność dzieci z tym nowotworem stopniowo rosła.
Epidemiologia |
Siatkówczak jest najczęstszym, pierwotnym, wewnątrzgałkowym, złośliwym guzem oka (siatkówki) u dzieci[17][18][19].
Częstość schorzenia szacuje się na 1:15 000–20 000 żywych urodzeń. W 60% przypadków guz jest jednostronny, a średnia wieku rozpoznania wynosi 2 lata. 15% tych guzów jest wrodzonych. Retinoblastoma jest obustronny w około 40% przypadków, a średni wiek rozpoznania wynosi wtedy 1 rok. Wszystkie obustronne i jednostronne wieloogniskowe guzy są wrodzone[20]. Odnotowano pewne różnice w zapadalności na ten typ nowotworu w różnych krajach. Częstość jest wyższa w krajach rozwijających się, w niektórych krajach Ameryki Środkowej i Południowej retinoblastoma jest najczęstszym litym guzem u dzieci[21]. Zapadalność w Polsce nie jest znana[19].
| Autor | Kraj | Okres badania | Częstość |
|---|---|---|---|
| Hemmes | Holandia | 1927-1929 | 1:34 000 |
| Griffith, Sorsby | Anglia | 1894-1943 | 1:32 793 |
| Falls, Neel[23] | Michigan, USA | 1938-1947 | 1:20 288 |
| Bohringer | Niemcy Zachodnie | 1925-1954 | 1:23 800 |
| Stevenson, Martin[24] | Irlandia Północna | 1938-1956 | 1:27 068 |
| Hemmes[25] | Holandia | 1952-1955 | 1:14 000 |
| Macklin[26] | Ohio, USA | 1940-1956 | 1:23 287 |
| Bech, Jensen[27] | Dania | 1928-1957 | 1:19 000 |
| Mork[28] | Norwegia | 1953-1960 | 1:17 000 |
| Schappert-Kimmijser[29] | Holandia | 1950-1959 | 1:15 230 |
| Suckling et al[30] | Nowa Zelandia | 1948-1968 | 1:17 500 |
| Barry, Mullaney[31] | Irlandia | 1955-1970 | 1:26 595 |
| Freedman, Goldberg[32] | Południowa Afryka (Bantu) | 1955-1975 | 1:10 000 |
| BenEzra, Chirambo[33] | Malawi | 1975 | 1:10 000 |
| Matsunaga, Ogyu[34] | Hokkaido, Japonia | 1945-1947 | 1:24 000 |
| Takano et al[35] | Nagasaki, Japonia | 1965-1986 | 1:16 000 |
Przyczyny zjawiska wyższej zapadalności na nowotwór w krajach rozwijających się nie są wyjaśnione. Prawdopodobnymi czynnikami są niski status socjoekonomiczny, niedobór kwasu foliowego w okresie ciąży i infekcja wirusem brodawczaka ludzkiego[36].
Opublikowano doniesienia, według których zapłodnienie in vitro (IVF) może wiązać się ze znacząco wyższym ryzykiem wystąpienia retinoblastoma u dziecka[37]. Badacze z ośrodka w Holandii, gdzie działa krajowy rejestr przypadków retinoblastoma, obliczyli ryzyko względne wystąpienia tego typu guza u grupy pacjentów urodzonych z IVF na 4,9–7,2[38]. Obliczenia oparte zostały na liczbie pięciu znanych przypadków występowania choroby u dzieci poczętych przy pomocy in vitro w latach 1995-2001 w Holandii. Badania zostały rozszerzone w 2009 roku poprzez analizę zachorowań dzieci urodzonych w latach 2002-2007, w których nie stwierdzono znacząco wyższego ryzyka zachorowania[39].
Patogeneza |

Schemat ilustrujący hipotezę Knudsona na przykładzie genu RB1
Retinoblastoma jest nowotworem uwarunkowanym genetycznie, związanym z mutacją antyonkogenu RB1 znajdującego się na chromosomie 13 w locus 13q14. Dziedziczy się autosomalnie dominująco z 90-95% penetracją. Paradoksalnie dodatni wywiad rodzinny występuje w około 6%, co spowodowane jest częstym spontanicznym pojawianiem się mutacji. U chorych z rodzinnym siatkówczakiem istnieje prawie 50% prawdopodobieństwo przekazania obciążającej mutacji potomstwu. Do wystąpienia choroby konieczna jest mutacja obu alleli, co zostało opisane przez amerykańskiego genetyka Alfreda Knudsona jako tzw. hipoteza dwóch uderzeń (ang. two hits)[15]. Przypadki rodzinne, w których jeden wadliwy allel jest dziedziczony stanowią około 40% stwierdzanych siatkówczaków; pozostałe 60% to przypadki sporadyczne, gdzie mutacje obu alleli zachodzą dopiero w czasie rozwoju osobniczego.
Objawy i przebieg |
| Objaw | Częstość |
|---|---|
| Leukokoria | 56% |
| Zez | 20% |
| Jaskra, bolesność, zaczerwienienie | 7% |
| Pogorszenie widzenia | 5% |
| Objawy widoczne w rutynowym badaniu okulistycznym | 3% |
| Jednostronne poszerzenie źrenicy | 2% |
Różnobarwność tęczówek | 2% |
| Wylew krwi do komory przedniej | 1% |
Oczopląs | 0,5% |
| Białe plamki na tęczówce | 0,5% |
| Utrata łaknienia, niechęć do ssania | 0,5% |

Siatkówczak w zaawansowanym stadium[41]
- Guz siatkówki
Najważniejszymi wczesnymi obawami guza siatkówki są leukokoria (biały refleks źreniczny) i zez. Leukokoria z początku jest niestała, widoczna nie pod każdym kątem i zależna od warunków oświetlenia podczas badania[20]. Niekiedy objaw widoczny jest na zdjęciach z lampą błyskową (wygląda inaczej niż zwykły efekt czerwonych oczu). Inne spotykane w siatkówczaku objawy to rubeoza tęczówki, wysięk ropny w przedniej komorze oka (rzekomy ropostek – pseudohypopyon) albo wylew krwi do przedniej komory oka (krwistek – hyphaema), woloocze (buphthalmia), zapalenie tkanki łącznej oczodołu i wytrzeszcz (exophthalmus). Niekiedy siatkówczak jest długo bezobjawowy[20]. Do rozsiewu nowotworu dochodzi za pośrednictwem naczyń krwionośnych błony naczyniowej. Guz nacieka poza gałkę oczną przez nerw wzrokowy i drogą przestrzeni podpajęczynówkowej. Może też naciekać twardówkę i szerzyć się w obrębie oczodołu. Przerzuty stwierdza się w węzłach chłonnych przedusznych i szyjnych[19]. Odległe przerzuty występują przede wszystkim do mózgu, kości czaszki i innych kości[19].
Objawy kliniczne retinoblastoma zależą od typu wzrostu guza, czasu choroby, stopnia waskularyzacji guza oraz obecności zwapnień, rozsiewu do ciała szklistego, odwarstwienia siatkówki lub krwotoku. Wyróżnia się kilka typów wzrostu guza:
typ endofityczny (endophytic growth pattern), gdy komórki guza dzielą się w obrębie wewnętrznych warstw siatkówki i gdy guz rośnie w kierunku szklistki
typ egzofityczny (exophytic growth pattern) ma miejsce, gdy guz rośnie z wewnętrznych warstw siatkówki poza siatkówkę do przestrzeni podsiatkówkowej (subretinal space), powodując jej odwarstwienie.
mieszany typ wzrostu polega na współwystępowaniu egzofitycznego i endofitycznego typu wzrostu guza.- wyjątkowo (2%), może naciekać płasko (plaquelike pattern) w sposób rozsiany, na powierzchni siatkówki lub poza nią, nie tworząc widocznego guza ani nawapnień wewnątrz, postępując powoli proksymalnie, objawiając się w późnym okresie choroby pod postacią rzekomozapalnych powikłań[42].
- Retinoma
Retinoma to niezłośliwy guz siatkówki, spotykany u pacjentów z mutacjami RB1. Ma postać uniesionego, szarego guzka siatkówki, otoczonego ogniskami proliferacji nabłonka barwnikowego siatkówki. Guz nie powiększa się i nie wiąże się z ryzykiem transformacji złośliwej. Sugerowano, że retinoma jest łagodną manifestacją mutacji w genie RB1 lub postacią regresji guza[43].
- Siatkówczak trójstronny
Określenie siatkówczaka trójstronnego odnosi się do synchronicznych guzów retinoblastoma w obu gałkach ocznych i szyszyniaka zarodkowego (pinealoblastoma) szyszynki lub okolicy nadsiodłowej. Szyszyniak jest guzem pochodzenia neuroektodermalnego przypominającym niskozróżnicowany guz typu retinoblastoma.
- Drugi nowotwór złośliwy (second malignancy)
Mutacje RB1 obecne we wszystkich komórkach pacjentów z siatkówczakiem predysponują ich też do innych niż siatkówczak nowotworów złośliwych, wykazujących predylekcję do określonych tkanek. Nowotwory te są główną przyczyną śmierci pacjentów, częstszą niż sam siatkówczak[44][45]. Kumulatywna częstość tych guzów szacowana jest na 1% na rok, co daje 50% ryzyko w wieku 50 lat[44]. Radioterapia znacznie zwiększa ryzyko nowotworzenia, zwłaszcza jeśli była stosowana w pierwszym roku życia[46]. Według badania Abramsona i wsp.[47] najczęstsze lokalizacje tych guzów to:
- tkanki miękkie głowy (24%)
- kości czaszki (18%)
- skóra (15%)
- mózg (8%)
- inne (25%).
Patomorfologia |

Obraz guza (strzałka) w wyłuszczonej gałce ocznej 3-letniej dziewczynki
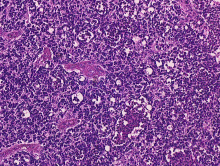
Siatkówczak w obrazie z mikroskopu świetlnego, barwienie H-E, 400-krotne powiększenie
Makroskopowo siatkówczak ma postać różowego lub białawego guzka z nawapnieniami, o typie wzrostu egzofitycznym, endofitycznym lub rozlanym, jedno- lub wieloogniskowym. Mikroskopowo retinoblastoma należy do guzów drobnookrągłoniebieskokomórkowych: zbudowany jest z niewielkich, niskozróżnicowanych, okrągłych komórek o okrągłym jądrze i wąskim paśmie cytoplazmy. Komórki guza przypominają niezróżnicowane retinoblasty, często są uchwycone w trakcie podziału mitotycznego. Między nimi zwykle są obecne liczne pola martwicy i dystroficznych zwapnień. Niekiedy komórki są bardziej zróżnicowane i tworzą wtedy układy rozetkowe, przede wszystkim tzw. rozetki Flexnera-Wintersteinera, rzadziej rozetki Homera Wrighta, typowe raczej dla neuroblastoma. Spotyka się też inne, tzw. kwiatopodobne formacje histologiczne (fleurette).
Histopatologiczne czynniki ryzyka |
Nie ma konsensusu odnośnie histopatologicznych czynników prognostycznych pozwalających przewidzieć przebieg choroby. Uważa się, że źle rokują:
- odległe zajęcie nerwu wzrokowego
- masywne nacieczenie naczyniówki
- rozrost pozatwardówkowy
- ogniskowe zajęcie naczyniówki
- rozsiew do komory przedniej
- rozsiew do ciała rzęskowego
- zajęcie tęczówki
- masywna martwica guza.
Rozpoznanie |

Obraz USG siatkówczaka u 3-letniego dziecka

Obraz siatkówczaka w fundoskopii[20]

Obraz MRI T1-zależny ze wzmocnieniem kontrastowym w projekcji strzałkowej[20]
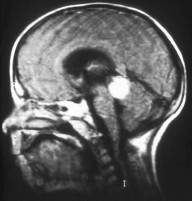
Obraz siatkówczaka trójstronnego w MRI głowy w projekcji strzałkowej
- Badanie dna oka
Badanie dna oka (fundoskopia) w znieczuleniu ogólnym i po maksymalnym farmakologicznym rozszerzeniu źrenicy umożliwia postawienie prawidłowej diagnozy. Badanie polega na starannym obejrzeniu przez okulistę całego dna oka aż po rąbek zębaty; zaleca się stosowanie ucisku na twardówkę w przedrównikowej części gałki[48]. W obrazie uzyskanym w fundoskopii widać białawy guzek z naczyniakowato poszerzonymi naczyniami[20]. Nawet bardzo małe guzy można wykryć wziernikowaniem w obrazie odwróconym z użyciem wziernika obuocznego.
- USG
W USG gałki ocznej siatkówczak prezentuje się jako masa o większej niż ciało szkliste echogeniczności, z drobnymi nawapnieniami. W przypadku guza o wzroście egzofitycznym można stwierdzić odwarstwienie siatkówki[20]. Badanie pozwala ocenić wielkość guza i zróżnicować go z naczyniakiem, ziarniniakiem albo toksokarozowym zapaleniem oka[48].
- MRI
MRI jest metodą z wyboru przy ocenie miejscowego rozrostu guza[20]. Intensywność sygnału z tkanki guza w obrazach T1-zależnych jest taka sama albo nieco większa jak ciała szklistego, a w obrazach T2-zależnych sygnał jest stosunkowo słabszy. W metodzie tej nie da się stwierdzić obecnych nawapnień w miąższu guza, można natomiast stwierdzić zajęcie nerwu wzrokowego, przedniej komory oka albo tkanki łącznej oczodołu. MRI pozwala zróżnicować siatkówczaka z takimi rzekomonowotworowymi zmianami, jak choroba Coatsa (zewnętrzne zapalenie siatkówki) albo wrodzone malformacje gałki ocznej i rozpoznać rzadkie przypadki siatkówczaka trójstronnego[49].
- Tomografia komputerowa
Tomografia komputerowa zazwyczaj wykazuje obecność wewnątrzgałkowej masy o większej niż ciało szkliste gęstości mierzonej w jednostkach Hounsfielda, posiadającej zwapnienia w 90% przypadków i ulegającej niewielkiemu wzmocnieniu kontrastowemu po podaniu dożylnego jodowego środka kontrastowego[20].
Testy genetyczne |
| Metoda | Rozpoznawane mutacje | Procent wykrywanych mutacji |
|---|---|---|
FISH | Submikroskopowe delecje i translokacje | >8% |
Badanie LOH | Submikroskopowe delecje i translokacje | 8% |
MLPA | Submikroskopowe delecje, insercje i rearanżacje | 16% |
qmPCR | Delecje, insercje | 37% |
Mutation scanning | Substytucje pojedynczych zasad, mutacje małej długości | 70-75% |
Sekwencjonowanie | Substytucje pojedynczych zasad, mutacje małej długości | 70-75% |
Targeted mutation analysis | Specyficzne, odziedziczone mutacje punktowe | 30% |
Methylation analysis | Hipermetylacja regionu promotorowego | 10-12% |
Badanie RNA | Mutacje miejsca splicingowego, duże rearanżacje | <5% |
Identyfikacja mutacji w genie RB1, która doprowadziła do siatkówczaka u dziecka, może być ważna w podejściu do opieki klinicznej nad pacjentem jak również w opiece nad (przyszłym) rodzeństwem i potomstwem.
- U pacjentów z obustronnym siatkówczakiem i u 13-15% pacjentów z jednostronną chorobą[51][52], spodziewana jest obecność RB1 mutacji we krwi. Dzięki identyfikacji mutacji RB1 u osoby dotkniętej siatkówczakiem, można tej mutacji szukać u (przyszłego) rodzeństwa, dzieci i innych krewnych. Jeśli nie posiadają oni tej mutacji, krewni dziecka nie są narażeni na siatkówczaka a więc nie muszą być poddawani urazom i kosztom badań pod narkozą[53]. Dla pacjentów z siatkówczakiem jednostronnym, u których w 85% przypadków nie znajduje się we krwi ani jednej z dwóch mutacji w RB1 znalezionych w ich nowotworze, badania molekularne ani nadzór medyczny ich rodzeństwa nie sa wymagane.
- Jeśli mutacja w RB1 jest zidentyfikowana u osoby dotkniętej siatkówczakiem, ta mutacja rodzinna może być poszukiwana w komórkach owodniowych w przypadku ciąży zagrożonej. Płód u którego zidentyfikuje się mutację może być poddany wcześniejszemu porodowi, co pozwala na wczesne rozpoczęcie leczenia nowotworów każdego oka, co z kolei prowadzi do lepszych rezultatów[53].
- W przypadku jednostronnego siatkówczaka, gdzie nowotwór oka nie jest dostępny do badań, jeśli mutacja RB1 nie została wykryta we krwi poprzez badania molekularne o dużej czułości (tj. badania które są zdolne wykryć ponad 93% mutacji RB1) ryzyko mutacji RB1 jest obniżone do poniżej 1%[52], poziom, przy którym tylko egzaminacja kliniczna (a nie badania pod narkozą) jest zalecana dla osoby dotkniętej chorobą i ich przyszłemu potomstwu[54].
Różnicowanie |
Diagnostyka różnicowa siatkówczaka obejmuje następujące choroby[55][56][57][58]:
- choroba Norriego
- wrodzone rozwarstwienie siatkówki
- zespół nietrzymania barwnika
- witreoretinopatia wysiękowa rodzinna
- przetrwałe hyperplastyczne pierwotne ciało szkliste
- anomalia tarczy nerwu wzrokowego w kształcie kwiatu powoju
- zaćma wrodzona
- wrodzony ubytek naczyniówki
- wrodzony fałd siatkówki
- wrodzone zmętnienie rogówki
toksokarozowe zapalenie gałki ocznej- toksoplazmoza
- cytomegalia
- zapalenie tkanek oczodołu
- gwiaździak
naczyniak naczyniówki- choroba Coatsa
- retinopatia wcześniaków
krwotok do ciała szklistego.
Leczenie |

Gordon Isaacs obok akceleratora liniowego, pierwszy pacjent poddany radioterapii z powodu siatkówczaka (1957)
Do niedawna jedynym sposobem leczenia było usunięcie zajętej gałki ocznej z przylegającym odcinkiem nerwu wzrokowego przed rozprzestrzenieniem się guza. W krajach rozwijających się wiele przypadków osiąga takim stadium zaawansowania, że dzieci umierają z powodu przerzutów.
Wybór metody leczenia i ewentualne kojarzenie terapii zależy od rozległości procesu nowotworowego i stanu drugiego oka. Zaproponowano różne algorytmy postępowania.
Wstępne leczenie chemioterapeutyczne (chemioredukcja) może zmniejszyć masę guza i umożliwić leczenie miejscowe[59].
Leczenie ogólnoustrojowe i chemioredukcja |
Chemioterapia jest bezwzględnie wskazana w przypadków zaawansowanych guzów (stwierdzony w tomografii komputerowej lub potwierdzony histopatologicznie naciek pozagałkowy błony naczyniowej lub nerwu wzrokowego poza blaszką sitową) oraz w przypadkach wystąpienia przerzutów. Wskazaniem względnym jest chemioredukcja[60]. Zaleca się chemioterapię wielolekową (karboplatyna, winkrystyna, etopozyd)[61]. Istotne jest przestrzeganie właściwego dawkowania w kolejnych cyklach z powodu ryzyka chemiooporności guzów. Niekiedy, w celu uniknięcia oporności wielolekowej, podaje się cyklosporynę A[62].

Schemat leczenia jednostronnego siatkówczaka według Aerts i wsp.[20]

Schemat leczenia obustronnego siatkówczaka według Aerts i wsp.[20]
Leczenie miejscowe |
Rodzaje leczenie miejscowego:
- krioterapia
- radioterapia
- fotokoagulacja laserowa
- termoterapia lub termochemioterapia
- miejscowa chemioterapia
enukleacja.
- Krioterapia
Krioterapię stosuje się w przypadku małych guzków, leżących na dalekim i bliskim obwodzie gałki ocznej. Polega na gwałtownym zamrożeniu guza, powodującym zniszczenie śródbłonka naczyń guza, zakrzepicę i zawał tkanki nowotworowej. Krioterapia wykonywana jest pod kontrolą wzroku i powtarzana trzykrotnie. Metoda ma mało powikłań i w 90% jest skuteczna wobec guzów o średnicy do 3 mm.
- Fotokoagulacja laserowa
Fotokoagulacja jest skuteczna w leczeniu małych zmian. W metodzie tej niszczy się guzy położone do tyłu od równika o średnicy podstawy do 10 mm i wysokości do 3 mm. Impulsy laserowe niszczą też unaczynienie guza.
- Przezźrenicza termo(chemio)terapia laserowa
Przezźrenicza termoterapia (transpupillary thermoterapy, TTT) polega na użyciu lasera diodowego i wiązki promieniowania laserowego zbliżonego do podczerwieni. Nagrzewanie guza prowokuje martwicę jego tkanek. Przed zabiegiem podaje się karboplatynę (termochemioterapia) i w mikroskopie operacyjnym przeprowadza się fotokoagulację masy guza laserem. Istnieje ryzyko zwiększenia rozsiewu komórek nowotworowych do ciała szklistego.
- Radioterapia
Radioterapia ma wysoką skuteczność w leczeniu siatkówczaka, ale obarczona jest poważnymi działaniami ubocznymi. Stosuje się radioterapię z pól zewnętrznych (akceleratorów – teleradioterapia) lub nadtwardówkowych aplikatorów (płytek) izotopowych (brachyterapia).
- Teleradioterapia
W tej metodzie stosuje się kolimatory, ogniskujące strumień o megawoltowej energii na szklistce. Obecnie wskazania do tej terapii ograniczone są do guzów zlokalizowanych w tylnym biegunie gałki ocznej, zwłaszcza w plamce i pęczku tarczkowo-plamkowym, a także dużych guzów (>10 mm). Ogólna dawka promieniowania wynosi 4000-4500 cGy na 4-6 tygodni leczenia.
Naświetlanie całej gałki ocznej stosuje się u jednoocznych pacjentów z nowotworem rozsianym do ciała szklistego. Powikłaniami leczenia są zaćma popromienna, neowaskularyzacja tęczówki i jaskra neowaskularyzacyjna. Poza tym, radioterapia z pól zewnętrznych skutkuje deformacjami twarzoczaszki, zaburzeniami endokrynologicznymi i neurokognitywnymi, a także zwiększonym ryzykiem wtórnych nowotworów w przyszłości (zobacz niżej). Zastosowanie skojarzonej chemioredukcji pozwala zmniejszyć dawkę promieniowania[potrzebny przypis].
- Brachyterapia
Jest to metoda kontaktowa, polegająca na naszywaniu na gałkę oczną aplikatorów z radioaktywnymi izotopami jodu, rutenu albo irydu. Aplikatory nadtwardówkowe zawierają izotopy: 106Ru, 125I, 192Ir. Pozostają one na oku przez 2-4 (5-7) dni, docelowo dostarczając 4.000-4.500 cGy przeztwardówkowo do guza. Wskazaniem do stosowania tej metody jest niepowodzenie innych metod zachowujących gałkę oczną; może to być leczenie pierwszego rzutu albo, częściej, drugiego rzutu u pacjentów nie odpowiadających na teleradioterapię. Efekty uboczne są rzadsze niż w tej ostatniej, wydaje się że ryzyko drugiego nowotworu nie istnieje. Guzy leczone brachyterapią mogą mieć największą średnicę podstawy do 16 mm i wysokość do 8 mm. Powikłaniami brachyterapii są neuropatia i waskulopatia popromienna.
- Chemioterapia miejscowa
Metoda znajduje zastosowanie u pacjentów z enukleowanym jednym okiem, u których wcześniej stosowane techniki leczenia nie spowodowały regresji. Przeprowadza się iniekcję chemioterapeutyku do ciała szklistego lub pod torebkę Tenona. Stosowane chemioterapeutyki to ThioTEPA i karboplatyna[63].
- Enukleacja
Wskazania do enukleacji (enucleatio) dzielą się na bezwzględne (gdy zabieg należy przeprowadzić bez względu na stan drugiego oka) oraz względne (gdy drugie oko jest zdrowe lub gdy proces chorobowy w drugim oku może być skutecznie leczony).
Do wskazań bezwzględnych należą:
- naciek pozagałkowy stwierdzony w tomografii komputerowej
- całkowite wypełnienie gałki ocznej z jaskrą wtórną i (lub) rozsiewem w komorze przedniej.
Do wskazań względnych należą:
- guz wypełniający ponad połowę gałki
- całkowite odwarstwienie siatkówki
- wznowa guza wzdłuż podstawy ciała szklistego
- rozsiew w ciele szklistym.
Enukleacja guza wymaga zmodyfikowanej techniki zabiegu, polegającej na zastosowaniu noża diatermicznego do odcięcia mięśni prostych i skośnych, pętli Fostera (umożliwia głębokie wejście do oczodołu i długie – około 10 mm – odcięcie nerwu wzrokowego) oraz wszczepów oczodołowych. Wszczepy z hydroksyapatytu lub porowatego polietylenu wypełniają oczodół i poprawiają ruchomość protezy, sprzyjają też prawidłowemu wzrostowi oczodołu u dzieci. Z czasem może rozwinąć się zespół poenukleacyjny oczodołu (ang. post-enucleation socket syndrome, PESS), polegający na retrakcji implantu i spłyceniu załamków szpary powiekowej, co skutkuje nieprawidłowym utrzymywaniem protezy. PESS jest wskazaniem do operacji rekonstrukcyjnej.
- Porównanie opcji terapeutycznych
| Terapia | Wskazania | Możliwe powikłania | Skuteczność |
|---|---|---|---|
| Termoterapia laserowa[65] | Jeden lub więcej małych guzów bez rozsiewu do ciała szklistego | Wznowa guza, odwarstwienie siatkówki, zator tętnicy siatkówki, włóknienie przedsiatkówkowe | Do 92% |
| Krioterapia[66] | Jeden lub więcej małych guzów bez rozsiewu do ciała szklistego; metoda nadaje się zwłaszcza dla guzów w przedniej części gałki ocznej | Przerwania siatkówki, odwarstwienie siatkówki, odwarstwienie naczyniówki, zapalenie jagodówki | 70% i więcej |
| Brachyterapia[67] | Guzy zbyt duże by zastosować termoterapię lub krioterapię | Neuropatia nerwu wzrokowego, retinopatia poradiacyjna | Do 90% |
| Teleradioterapia[68] | Duże obustronne guzy nie poddające się chemioterapii; guzy w sąsiedztwie dołka środkowego lub nerwu wzrokowego; guzy zbyt duże by zastosować termoterapię lub krioterapię. Metoda nie powinna być stosowana przed ukończeniem 1. roku życia | Neuropatia nerwu wzrokowego, retinopatia radiacyjna, zaćma, zespół suchego oka, atrofia mięśniowa; wysokie ryzyko nowotworów wtórnych | Do 90% |
| Chemioterapia[69] | Podejrzenie lub potwierdzenie przerzutów guza; redukcja rozmiarów guza przed rozpoczęciem leczenia miejscowego | Supresja szpiku kostnego, nefropatia, głuchota, ryzyko białaczki | 50-90% |
| Enukleacja[70] | Jednostronne, duże guzy zajmujące 50% i więcej objętości gałki ocznej, przebiegające z rozrostem w kierunku przednim lub jaskrą neowaskularyzacyjną, albo nie odpowiadające na leczenie zachowawcze | W zasadzie bezpieczna, rzadko powikłania: krwotok lub zakażenie | 95% |
Terapie eksperymentalne |
- Terapia fotodynamiczna
Terapia fotodynamiczna (photodynamic therapy, PDT), oparta na użyciu nietoksycznych fotouczulaczy aktywowanych światłem niejonizującego lasera, może stanowić nową opcję terapeutyczną. Pierwsze eksperymentalne dane wykazują, że PDT może w istotny sposób spowolnić wzrost guza[71].
- Terapia genowa
Na etapie badań klinicznych są próby terapii genowej w siatkówczaku z użyciem wektora adenowirusowego[72].
Regresja guza |
Wyróżnia się pięć typów regresji guza[73]:
Typ 0 – całkowity zanik guza
Typ I – pozostałości guza za sprawą ognisk zwapnień mają obraz tzw. pokruszonego sera (ang. cottage cheese appearance)
Typ II – szara, półprzejrzysta tkanka guza, tzw. obraz rybiego mięsa (ang. fish flesh appearance)
Typ III (mieszany) – łączy cechy typów I i II
Typ IV – atrofia siatkówki, naczyniówki i guza, najczęściej po krioterapii lub TCT.
Rokowanie |
Opracowano kilka klasyfikacji klinicznych wewnątrzgałkowego siatkówczaka, z których dwie: klasyfikacja Reese'a i Ellswortha oceniająca szansę zachowania oka w przypadku radioterapii guza i klasyfikacja ABC, oceniająca szansę zachowania oka przy wdrożeniu nowszych terapii, używane są najczęściej[20]. Niedawno zaproponowano nową klasyfikację (International Retinoblastoma Classification) oceniającą rokowanie w przypadku całego spektrum choroby, także guzów naciekających poza oczodół.
| Grupa | Opis |
|---|---|
| I | a) pojedynczy guz wielkości < 4-krotnej średnicy tarczy nerwu wzrokowego, w równiku lub poza równikiem gałki ocznej b) mnogie guzy, żaden z nich nie ma wielkości > od 4-krotnej średnicy tarczy nerwu wzrokowego, w równiku gałki ocznej lub poza nim |
| II | a) pojedynczy guz, o średnicy 4–10-krotnie większej od średnicy tarczy nerwu wzrokowego, w równiku lub poza równikiem gałki ocznej b) mnogie guzy, o średnicy 4–10-krotnie większej od średnicy tarczy nerwu wzrokowego, poza równikiem gałki ocznej |
| III | a) zmiana każdej wielkości położona do przodu od równika gałki ocznej b) pojedynczy guz wielkości > 10-krotnej średnicy tarczy nerwu wzrokowego, poza równikiem gałki ocznej |
| IV | a) liczne guzy, niektóre z nich przekraczają > 10-krotnie średnicę tarczy nerwu wzrokowego b) każda zmiana szerząca się do przodu od rąbka zębatego (ora serrata) |
| V | a) duże guzy zajmujące więcej niż połowę powierzchni siatkówki b) rozsiew do ciała szklistego |
| Grupa | Opis |
|---|---|
| A | Małe guzy oddalone od dołka środkowego siatkówki i tarczy nerwu wzrokowego; guzy < 3 mm średnicy w najszerszym wymiarze ograniczone do siatkówki i zlokalizowane przynajmniej 3 mm od dołka środkowego i 1,5 mm od tarczy nerwu wzrokowego |
| B | Wszystkie pozostałe guzy ograniczone do siatkówki; wszystkie guzy ograniczone do siatkówki i nie należące do grupy A, podsiatkówkowe nagromadzenie płynu (bez rozsiewu poniżej siatkówki) < 3 mm od podstawy guza |
| C | Miejscowe nagromadzenie płynu poniżej siatkówki lub rozsiew do ciała szklistego; podsiatkówkowy płyn 3–6 mm od podstawy guza lub rozsiew do ciała szklistego albo poniżej siatkówki na odległość < 3 mm od guza |
| D | Rozlane nagromadzenie podsiatkówkowe płynu albo rozsiew nowotworu; płyn > 6 mm od guza albo rozsiew do ciała szklistego lub podsiatkówkowy > 3 mm od guza |
| E | Obecność przynajmniej jednej źle rokującej cechy: więcej niż 2/3 gałki ocznej zajęte przez masę guza, guz w przedniej połowie gałki ocznej albo do przodu od ciała szklistego; guz w lub na ciele rzęskowym; neowaskularyzacja tęczówki (rubeoza); jaskra neowaskularyzacyjna; krwotok; martwica guza z aseptycznym zapaleniem oczodołu; phthisis bulbi oculi |
| Grupa | Opis |
|---|---|
| 0 | Pacjenci leczeni zachowawczo (klasyfikowani według skal oftalmologicznych przedoperacyjnych) |
| I | Oko wyłuszczone, histologicznie potwierdzona doszczętna resekcja |
| II | Oko wyłuszczone, histologicznie potwierdzona niedoszczętność resekcji |
| III | Regionalne szerzenie się guza: a) jawne zajęcie oczodołu b) zajęcie węzłów chłonnych przedusznych lub szyjnych |
| IVa | Przerzuty nowotworu drogą naczyń krwionośnych 1. pojedynczy przerzut 2. mnogie przerzuty |
| IVb | Szerzenie się przez ciągłość do ośrodkowego układu nerwowego 1. zmiany przed skrzyżowaniem wzrokowym 2. masa w ośrodkowym układzie nerwowym 3. zajęcie opon mózgowych |
Przeżywalność w przypadku samego siatkówczaka jest obecnie doskonała u pacjentów z jednostronnym lub obustronnym guzem; w krajach zachodnich wskaźnik wyleczeń wynosi około 95%[20]. Zachowanie widzenia w zajętym oku zależy od metody leczenia, objętości guza w momencie rozpoznania, relacji anatomicznej nowotworu do plamki żółtej i nerwu wzrokowego oraz wystąpienia ewentualnych efektów niepożądanych leczenia (zaćma, krwotok do ciała szklistego). Na wieloletnie przeżycie w dziedzicznej postaci siatkówczaka mają wpływ inne nowotwory związane z mutacjami w genie RB1[77][78]. Niedawne badania wykazały, że po 50 latach od zdiagnozowania siatkówczaka inne nowotwory rozwijają się u 36% pacjentów z dziedziczną i u 5,7% pacjentów z niedziedziczną postacią guza[79]. Leczenie pacjentów z dziedzicznym siatkówczakiem może zwiększać ryzyko wystąpienia nowotworów w innych niż gałka oczna lokalizacjach, przez indukcję utraty heterozygotyczności w innych tkankach organizmu. Stwierdzono że teleradioterapia u niemowląt i chemioterapia mogą mieć odległe następstwa w postaci wystąpienia wtórnych nowotworów[80][81]. Najczęściej rozwijają się kościopochodne mięsaki czaszki i kości długich, mięsaki tkanek miękkich, czerniak złośliwy skóry, guzy mózgu, raki sutka i płuc.
Nowe kierunki badań |

Mysi model siatkówczaka; gen Rb transgenicznej myszy Chx10-Cre został inaktywowany w komórkach progenitorowych siatkówki[82]
Retinoblastoma jest wyłącznie chorobą ludzi; nie ma naturalnego modelu zwierzęcego tego nowotworu[83]. Przy użyciu linii komórkowych otrzymanych z guzów człowieka wprowadzonych do ciała szklistego oka myszy (model przeszczepiania w układzie ksenogenicznym) otrzymano użyteczne do badań nad możliwościami leczniczymi nowotworu gryzonie[84][85][86]. Opracowano także transgeniczne mysie modele choroby[87][88]. Obecnie badania skupiają się na opracowaniu terapii genowej nowotworu oraz na minimalizacji skutków ubocznych opracowanych już i stosowanych metod leczenia.
Poradnictwo genetyczne i profilaktyka |
Dostępne są testy genetyczne pozwalające na wykrycie lub wykluczenie mutacji RB1 u krewnych probanta i tym samym wyłączenie ich z algorytmu postępowania profilaktycznego dla osób ze zwiększonym ryzykiem nowotworu. Wczesne wykrycie guza u objętych programem profilaktyki osób znacznie poprawia wyniki leczenia. Do chwili wykluczenia predyspozycji genetycznych członkowie rodziny pacjenta są traktowani jako potencjalni nosiciele mutacji RB1.
| Prezentacja guza u probanda | Wywiad rodzinny | Ryzyko choroby u rodzeństwa | Ryzyko choroby u potomstwa |
|---|---|---|---|
| Guz obustronny | Negatywny | 2%[89] | ≥50% |
| Guz jednostronny wieloogniskowy | Negatywny | 1-2%[89] | 6-50% |
| Guz jednostronny jednoogniskowy | Negatywny | 1% | 2-6% |
| Guz jednostronny jednoogniskowy | Pozytywny | Zmienne[a] | Zmienne[a] |
| Guz obustronny | Pozytywny | 50% | 50% |
Uwagi |
↑ ab W rodzinach z jednostronnym siatkówczakiem penetracja jest bardzo zmienna.
Przypisy |
↑ Young J.L., Smith M.A., Roffers S.D. i in. Retinoblastoma. W: Cancer Incidence and Survival among Children and Adolescents: United States SEER Program, 1975-1995, Ries L.A., Smith M.A., Gurney J.G. i in., National Cancer Institute, Bethesda, Maryland 1999. s. 73.
↑ ab Observatio XXIII. Tumor Oculorum. Observatories Anatomicae Selectiores. W: Pieter Pauw: Historiarum Anatomicarum Rariorum, Centuria III & IV. Kopenhaga: Petrus Morsing, 1657, s. 38-39.
↑ ab Wardrop, J: Observations on the fungus haematodes. Edinburgh: Constable, 1809.
↑ Abramson, DH. Retinoblastoma in the 20th Century: Past Success and Future Challenges The Weisenfeld Lecture. „Investigative Ophthalmology and Visual”. 46, s. 2684-2691, 2005. PMID: 16043839.
↑ Kivelä, T, Polkunen, ML. Pieter Pauw's Tumor Oculorum. Reappraisal of the Presumed First Description of Retinoblastoma in 1597. „Arch Ophthalmol”. 121 (6), s. 881-886, 2003. PMID: 12796262.
↑ Dunphy, EB. The story of retinoblastoma. „Trans Am Acad Ophthalmol Otolaryngol”. 68, s. 249-265, 1964. PMID: 14218388.
↑ Virchow, R: Die Drankhaften Geschwulste. 1864.
↑ Flexner S. A peculiar glioma (neuroepithelioma) of the retina. „Bull Johns Hopkins Hosp”. 2, s. 115-119, 1891.
↑ Wintersteiner H: Die Neuroepithelioma Retinae: Eine Anatomiche und Klnische Studie. Lipsk: Dentisae, 1897.
↑ Verhoeff, FH. Retinoblastoma. „Trans Am Ophthalmol Soc”. 24, s. 38-41, 1929.
↑ Ts'o MO, Zimmerman LE, Fine BS, Ellsworth RM. A cause of radioresistance in retinoblastoma: photoreceptor differentiation. „Trans Am Acad Ophthalmol Otolaryngol”. 74 (5), s. 959–969, 1970. PMID: 4990220.
↑ Stallard HB. The conservation treatment of retinoblastoma. „Trans Ophthal Soc”. 82, s. 473, 1962.
↑ Lele KP, Penrose LS, Stallard HB. Chromosome deletion in a case of retinoblastoma. „Ann Hum Genet”. 27, s. 171-174, 1963. PMID: 14081487.
↑ Orye E, Delbeke MJ, Vandenabeele B. Retinoblastoma and D-chromosome deletions (Letter). „Lancet II”. 1376, 1971.
↑ ab Knudson A. Mutation and cancer: statistical study of retinoblastoma. „Proc Natl Acad Sci U S A”. 68. 4, s. 820-823, 1971. PMID: 5279523.
↑ Hilgartner, HL. Report of a case of double glioma treated by x-ray. „Texas Med J”. 18, s. 322–323, 1903. PMID: 16152902.
↑ DH. Abramson, AC. Schefler. Update on retinoblastoma. „Retina”. 24 (6), s. 828-848, 2004. DOI: 10.1097/00006982-200412000-00002. PMID: 15579980.
↑ Castillo, BV, Kaufman, L. Pediatric tumors of the eye and orbit. „Pediatr Clin North Am”. 50. 1, s. 149-172, 2003. PMID: 12713110.
↑ abcd Jerzy Stachura, Wenancjusz Domagała: Patologia znaczy słowo o chorobie. Kraków: Wydawnictwo PAU, 2003, s. 422-424. ISBN 83-88857-65-7.
↑ abcdefghijklm Aerts, I, Lumbroso-Le Rouic, L, Marion Gauthier-Villars, M, Brisse, H, Doz, F, Desjardins, L. Retinoblastoma. „Orphanet Journal of Rare Diseases”. 1. 31, 2006. PMID: 16934146.
↑ Leal-Leal C, Flores-Rojo M, Medina-Sansón A, Cerecedo-Díaz F, Sánchez-Félix S, González-Ramella O, Pérez-Pérez F, Gómez-Martínez R, Quero-Hernández A, Altamirano-Alvarez E, Alejo-González F, Figueroa-Carbajal J, Ellis-Irigoyen A, Tejocote-Romero I, Cervantes-Paz R, Pantoja-Guillén F, Vega-Vega L, Carrete-Ramírez F. A multicentre report from the Mexican Retinoblastoma Group. „Br J Ophthalmol”. 88, s. 1074–1077, 2004. PMID: 15258028.
↑ Bishop, JO, Madsen, EC. Retinoblastoma: a review of current status. „Surv Ophthalmol”. 19, s. 342-366, 1975. PMID: 1145423.
↑ Falls, HF, Neel, JV. Genetics of retinoblastoma. „AMA Arch Ophthalmol”. 46. 4, s. 367-389, 1951. PMID: 14868057.
↑ Stevenson, AC, Martin, VA. Retinoblastoma; occurrence of the condition in Northern Ireland, 1938-1956. „Br J Prev Soc Med”. 11 (1), s. 29-35, 1957. PMID: 13413175.
↑ Hemmes, GD. Morbidity & prognosis of retinoblastoma (glioma retinae) in the Netherlands. „Geneeskd Bl”. 48. 9, s. 1-29, 1958. PMID: 13548489.
↑ Macklin, MT. Inheritance of retinoblastoma in Ohio. „Arch Ophthalmol”. 62, s. 842-51, 1959. PMID: 14419546.
↑ Bech, K, Jensen, OA. Bilateral retinoblastoma in Denmark, 1928-1957. „Acta Ophthalmol (Kopenhaga)”. 39, s. 561-568, 1961. PMID: 13866234.
↑ Mork, T. Malignant neoplasms of the eye in Norway. Incidence, treatment and prognosis. „Acta Ophthalmol (Kopenhaga)”. 39, s. 824-831, 1961. PMID: 14476168.
↑ Schappert-Kimmijser, J. Retinoblastoma problems in the Netherlands. „Ophthalmologica”. 163. 1, s. 12-14, 1971. PMID: 5131968.
↑ Suckling, RD, Fitzgerald, PH, Stewart, J, Wells, E. The incidence and epidemiology of retinoblastoma in New Zealand: A 30-year survey. „Br J Cancer”. 46. 5, s. 729-36, 1982. PMID: 7171454.
↑ Barry, G, Mullaney, J. Retinoblastoma in the Republic of Ireland (1955-70). „Trans Ophthalmol Soc U K”. 91, s. 839-55, 1971. PMID: 5291569.
↑ Freedman, J, Goldberg, L. Incidence of retinoblastoma in the Bantu of South Africa. „Br J Ophthalmol”. 60. 9, s. 655-656, 1976. PMID: 990237.
↑ BenEzra, D, Chirambo, MC. Incidence of retinoblastoma in Malawi. „J Pediatr Ophthalmol”. 13. 6, s. 340-3, 1976. PMID: 1018220.
↑ Matsunaga, E, Ogyu. Genetic study of retinoblastoma in a Japanese population. „Jpn J Human Genet”. 4, s. 156, 1959.
↑ Takano, J, Akiyama, K, Imamura, N, Sakuma, M, Amemiya, T. Incidence of retinoblastoma in Nagasaki Prefecture, Japan. „Ophthalmic Paediatr Genet”. 12. 3, s. 139-44, 1991. PMID: 1754161.
↑ Orjuela M, Castaneda VP, Ridaura C, Lecona E, Leal C, Abramson DH, Orlow I, Gerald W, Cordon-Cardo C. Presence of human papilloma virus in tumor tissue from children with retinoblastoma: An alternative mechanism for tumor development. „Clin Cancer Res”. 6, s. 4010–4016, 2000. PMID: 11051250.
↑ BenEzra, D. In-vitro fertilisation and retinoblastoma. „Lancet”. 361, s. 273-274, 2003. PMID: 12559858.
↑ Moll AC, Imhof SM, Cruysberg JR, Schouten-van Meeteren AY, Boers M, van Leeuwen FE. Incidence of retinoblastoma in children born after in-vitro fertilisation. „Lancet”. 361. 9354, s. 309-10, 2003. PMID: 12559867.
↑ T. Marees. Incidence of retinoblastoma in Dutch children conceived by IVF: an expanded study. „Human Reproduction”. 24 (12), s. 3220–3224, 2009-09-25. ESHRE.
↑ Ellsworth RM. The practical management of retinoblastoma. „Trans Am Ophthalmol Soc”. 67, s. 462-534, 1969. PMID: 5381307.
↑ Oram Ring, G. Recurrent Neuroepithelioma. „Trans Am Ophthalmol Soc”. 15, s. 217–226, 1917. PMID: 1318121.
↑ Balmer, A, Zografos, L, Munier, F. Diagnosis and current management of retinoblastoma. „Oncogene”. 25. 38, s. 5341-5349, 2006. DOI: 10.1038/sj.onc.1209622. PMID: 16936756.
↑ Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: spontaneous regression of retinoblastoma or benign manifestation of the mutation?. „Brit J Cancer”. 45, s. 513-521, 1982. PMID: 7073943.
↑ ab Abramson, DH. Second nonocular cancers in retinoblastoma: a unified hypothesis. The Franceschetti Lecture. „Ophthalmic Genet”. 20. 3, s. 193-204, 1999. PMID: 10610188.
↑ Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. „J Natl Cancer Inst”. 96. 5, s. 357-63, 2004. PMID: 14996857.
↑ Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. „Ophthalmology”. 105. 4, s. 573-579, 1998. PMID: 9544627.
↑ Abramson DH, Melson MR, Dunkel IJ, Frank CM. Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. „Ophthalmology”. 108. 10, s. 1868-76, 2001. PMID: 11581064.
↑ ab Okulistyka. Podstawy kliniczne. Maria Hanna Niżankowska (red.). Warszawa: Wydawnictwo Lekarskie PZWL, 2007, s. 416-420. ISBN 978-83-200-3224-6.
↑ Brisse HJ, Lumbroso L, Freneaux PC, Validire P, Doz FP, Quintana EJ, Berges O, Desjardins LC, Neuenschwander SG. Sonographic, CT, and MR imaging findings in diffuse infiltrative retinoblastoma: report of two cases with histologic comparison. „AJNR Am J Neuroradiol”. 22, s. 499-504, 2001. PMID: 11237973.
↑ ab Dietmar R. Lohmann, Brenda L. Gallie: Retinoblastoma. Gene Reviews, 18-7-2000. [dostęp 2012-02-13].
↑ Schüler A, Weber S, Neuhäuser M,, et al.. Age at diagnosis of isolated unilateral retinoblastoma does not distinguish patients with and without a constitutional RB1 gene mutation but is influenced by a parent-of-origin effect. „Eur J Cancer”. 5. 41, s. 735–40, 2005. PMID: 15763650.
↑ ab Rushlow D, Piovesan B, Zhang K, et al.. Detection of mosaic RB1 mutations in families with retinoblastoma.. „Hum Mutat”. 5. 30, s. 842–51, 2009. PMID: 19280657.
↑ ab Richter S, Vandezande K, Chen N, et al.. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. „Am J Hum Genet”. 2. 72, s. 253–69, 2002. PMID: 12541220.
↑ Canadian Ophthalmological Society. National Retinoblastoma Strategy Canadian Guidelines for Care; Genetic Analysis. „Canadian Journal of Ophthalmology”. Suppl2. 44, s. S17–S22, 2009. PMID: 20237571. [zarchiwizowane z adresu].
↑ Kaufman LM, Mafee MF, Song CD. Retinoblastoma and simulating lesions. Role of CT, MR imaging and use of Gd-DTPA contrast enhancement. „Radiol Clin North Am”. 36, s. 1101-1117, 1998. PMID: 9884691.
↑ Meier P, Sterker I, Tegetmeyer H. Leukokorie im Kindesalter. „Klin Monatsbl Augenheilkd”. 223. 6, s. 521-527, 2006. DOI: 10.1055/s-2005-859005. PMID: 16804823.
↑ Shields JA, Parsons HM, Shields CL, Shah P. Lesions simulating retinoblastoma. „J Pediatr Ophthalmol Strabismus”. 28. 6, s. 338-40, 1991. PMID: 1757860.
↑ Shields JA, Shields CL, Parsons HM. Differential diagnosis of retinoblastoma. „Retina”. 11. 2, s. 232-43, 1991. PMID: 1925090.
↑ Dunkel IJ, Lee TC, Shi W, Beaverson KL, Novetsky D, Lyden D, Finlay JL, McCormick B, Abramson DH. A phase II trial of carboplatin for intraocular retinoblastoma. „Pediatr Blood Cancer”, 2005. PMID: 17301956.
↑ Shields C.L., Shields J.A., De Potter P. New treatment modalities for retinoblastoma. „Current Opinion in Ophthalmology”. 3, s. 20-26, 1996. PMID: 10163455.
↑ White L: Chemotherapy for retinoblastoma. „Med Paediatr Oncol”. 24, s. 341-342, 1995. PMID: 7700189.
↑ Chan H.S., Gallie B.L., Munier F.L., Beck Popovic M. Chemotherapy for retinoblastoma. „Ophthalmol Clin North Am”. 18, s. 55–63, 2005. PMID: 15763191.
↑ Abramson DH, Frank CM, Dunkel IJ. A phase I/II study of subconjunctival carboplatin for intraocular retinoblastoma. „Ophthalmology”. 106, s. 1947-1950, 1999. DOI: 10.1016/S0161-6420(99)90406-2. PMID: 10519590.
↑ Melamud A, Palekar R, Singh A. Retinoblastoma. „Am Fam Physician”. 73. 6, s. 1039-44, 2006. PMID: 16570739.
↑ Abramson DH, Schefler AC. Transpupillary thermotherapy as initial treatment for small intraocular retinoblastoma: technique and predictors of success. „Ophthalmology”. 111, s. 984-991, 2004. DOI: 10.1016/j.ophtha.2003.08.035. PMID: 15121378.
↑ Abramson DH, Ellsworth RM, Rozakis GW. Cryotherapy
for retinoblastoma. „Arch Ophthalmol”. 100, s. 1253-1256, 1982. PMID: 7103809.
↑ Shields JA, Shields CL, De Potter P, Hernandez JC,
Brady LW. Plaque radiotherapy for residual or recurrent retinoblastoma in 91 cases. „J Pediatr Ophthalmol Strabismus”. 31, s. 242-245, 1994. PMID: 7807301.
↑ Blach LE, McCormick B, Abramson DH. External beam radiation therapy and retinoblastoma: long-term results in the comparison of two techniques. „Int J Radiat Oncol Biol Phys”. 35, s. 45-51, 1996. DOI: 10.1016/S0360-3016(96)85010-3. PMID: 8641925.
↑ Murphree AL, Villablanca JG, Deegan WF 3rd, Sato JK, Malogolowkin M, Fisher A, Parker R, Reed E, Gomer CJ. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. „Arch Ophthalmol”. 114, s. 1348-56, 1996. PMID: 8906025.
↑ Shields JA, Shields CL, Sivalingam V. Decreasing frequency of enucleation in patients with retinoblastoma. „Am J Ophthalmol”. 108, s. 185-188, 1989. PMID: 2757099.
↑ Aerts I, Leuraud P, Laville I, Blais J, Maillard P, Desjardins L
et al. „International Retinoblastoma Symposium. International Society of Genetic Eye Disease and International Retinoblastoma Symposium: Whistler, Canada”, 2005.
↑ Chévez-Barrios P, Chintagumpala M, Mieler W, Paysse E, Boniuk M, Kozinetz C, Hurwitz MY, Hurwitz RL. Response of retinoblastoma with vitreous tumor seeding to adenovirus-mediated delivery of thymidine kinase followed by ganciclovir. „J Clin Oncol”. 23. 31, s. 7927-7935, 2005. DOI: 10.1200/JCO.2004.00.1883. PMID: 16258092.
↑ Singh AD, Garway-Heath D, Love S, Plowman PN, Kingston JE, Hungerford JL. Relationship of regression pattern to recurrence in retinoblastoma. „Br J Ophthalmol”. 77. 1, s. 12-16, 1993. DOI: 10.1136/bjo.77.1.12. PMID: 8435390.
↑ Reese AB, Ellsworth R. The evaluation and current concept of retinoblastoma therapy. „Trans Am Acad Ophthalmol Otolaryngol”. 67, s. 164–172, 1963. PMID: 13973597.
↑ Murphree, AL. Intraocular Retinoblastoma: the Case for a New Group Classification. „Ophthalmol Clin North Am”. 18, s. 41-53, 2005. PMID: 15763190.
↑ Chantada, G, Doz, F, Antoneli, CB, Grundy, R, Clare Stannard, FF, Dunkel, IJ, Grabowski, E, Leal-Leal, C, Rodriguez-Galindo, C, Schvartzman, E, Popovic, MB, Kremens, B, Meadows, AT, Zucker, JM. A proposal for an international retinoblastoma staging system. „Pediatr Blood Cancer”. 47. 6, s. 801-805, 2006. PMID: 16358310.
↑ Aerts, I, Pacquement, H, Doz, F, Mosseri, V, Desjardins, L, Sastre, X, Michon, J, Rodriguez, J, Schlienger, P, Zucker, JM, Quintana, E. Outcome of second malignancies after retinoblastoma: a retrospective analysis of 25 patients treated at the Institut Curie. „Eur J Cancer”. 40, s. 1522-1529, 2004. PMID: 15196536.
↑ Eng, C, Li, FP, Abramson, DH, Ellsworth, RM, Wong, FL, Goldman, MB, Seddon, J, Tarbell, N, Boice, JD Jr. Mortality from second tumors among long-term survivors of retinoblastoma. „J Natl Cancer Inst”. 85, s. 1121-1128, 1993. PMID: 8320741.
↑ Kleinerman, RA, Tucker, MA, Tarone, RE, Abramson, DH, Seddon, JM, Stovall, M, Li, FP, Fraumeni, JF Jr. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. „J Clin Oncol”. 23 (10), s. 2272-2279, 2005. PMID: 15800318.
↑ Abramson DH, Frank CM: Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. „Ophthalmology”. 105, s. 573-579, 1998.
↑ Wong, FL, Boice, JD Jr, Abramson, DH, Tarone, RE, Kleinerman, RA, Stovall, M, Goldman, MB, Seddon, JM, Tarbell, N, Fraumeni, JF Jr, Li, FP. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. „JAMA”. 278 (15), s. 1262-1267, 1997. PMID: 9333268.
↑ Michael A. Dyer, Carlos Rodriguez-Galindo, Matthew W. Wilson. Use of Preclinical Models to Improve Treatment of Retinoblastoma. „PLoS Med”. 2. 10. s. e332. DOI: 10.1371/journal.pmed.0020332. PMID: 16231976.
↑ Chintagumpala M, Chevez-Barrios P, Paysse EA, Plon SE, Hurwitz R. Retinoblastoma: Review of Current Management. „Oncologist”. 12. 10, s. 1237-1246, 2007. DOI: 10.1634/theoncologist.12-10-1237. PMID: 17962617.
↑ Gallie BL, Albert DM, Wong JJ et al. Heterotransplantation of retinoblastoma into the athymic "nude" mouse. „Invest Ophthalmol Vis Sci”. 16, s. 256–259, 1977.
↑ Rowe SG, Lee WH, Madreperla S. Subretinal and vitreal growth of human retinoblastoma cells in the mouse eye. „Invest Ophthalmol Vis Sci”. 33, s. 875, 1992.
↑ del Cerro M, Seigel GM, Lazar E et al. Transplantation of Y79 cells into rat eyes: An in vivo model of human retinoblastomas. „Invest Ophthalmol Vis Sci”. 34, s. 3336–3346, 1993.
↑ Albert DM, Griep AE, Lambert PF et al. Transgenic models of retinoblastoma: What they tell us about its cause and treatment. „Trans Am Acad Ophthalmol Soc”. 92, s. 385–400, 400–401, 1994. PMID: 1298518.
↑ Howes KA, Lasudry JG, Albert DM, Windle JJ. Photoreceptor cell tumors in transgenic mice. „Invest Ophthalmol Vis Sci”. 35, s. 342–351, 1994. PMID: 8112979.
↑ ab Jeżeli rodzeństwo jest zdrowe; zobacz Draper GJ, Sanders BM, Brownbill PA, Hawkins MM. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. „Br J Cancer”. 66, s. 211-9, 1992. PMID: 1637670.
Linki zewnętrzne |
Retinoblastoma; RB1 w bazie Online Mendelian Inheritance in Man (ang.)
- Marichelle L Aventura: Retinoblastoma. eMedicine Ophthalmology.
Retinoblastoma International (ang.)
http://www.cancer.gov/cancertopics/types/retinoblastoma Retinoblastoma] na stronie National Cancer Institute (ang.)
Retinoblastoma Genetics (ang.)
Retinoblastoma. [zarchiwizowane z tego adresu]. na stronie NIH/UW GeneTests (ang.). [zarchiwizowane z tego adresu].
![]() Zapoznaj się z zastrzeżeniami dotyczącymi pojęć medycznych i pokrewnych w Wikipedii.
Zapoznaj się z zastrzeżeniami dotyczącymi pojęć medycznych i pokrewnych w Wikipedii.

